Description: Homo sapiens histone cluster 1, H2aj (H2AC14), mRNA. RefSeq Summary (NM_021066): Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Two molecules of each of the four core histones (H2A, H2B, H3, and H4) form an octamer, around which approximately 146 bp of DNA is wrapped in repeating units, called nucleosomes. The linker histone, H1, interacts with linker DNA between nucleosomes and functions in the compaction of chromatin into higher order structures. This gene is intronless and encodes a replication-dependent histone that is a member of the histone H2A family. Transcripts from this gene lack polyA tails but instead contain a palindromic termination element. This gene is found in the small histone gene cluster on chromosome 6p22-p21.3. [provided by RefSeq, Aug 2015]. Transcript (Including UTRs) Position: hg19 chr6:27,782,080-27,782,518 Size: 439 Total Exon Count: 1 Strand: - Coding Region Position: hg19 chr6:27,782,132-27,782,518 Size: 387 Coding Exon Count: 1
ID:H2A1J_HUMAN DESCRIPTION: RecName: Full=Histone H2A type 1-J; AltName: Full=Histone H2A/e; FUNCTION: Core component of nucleosome. Nucleosomes wrap and compact DNA into chromatin, limiting DNA accessibility to the cellular machineries which require DNA as a template. Histones thereby play a central role in transcription regulation, DNA repair, DNA replication and chromosomal stability. DNA accessibility is regulated via a complex set of post-translational modifications of histones, also called histone code, and nucleosome remodeling. SUBUNIT: The nucleosome is a histone octamer containing two molecules each of H2A, H2B, H3 and H4 assembled in one H3-H4 heterotetramer and two H2A-H2B heterodimers. The octamer wraps approximately 147 bp of DNA. SUBCELLULAR LOCATION: Nucleus. Chromosome. PTM: The chromatin-associated form is phosphorylated on Thr-121 during mitosis (Probable). PTM: Deiminated on Arg-4 in granulocytes upon calcium entry. PTM: Monoubiquitination of Lys-120 (H2AK119Ub) by RING1 and RNF2/RING2 complex gives a specific tag for epigenetic transcriptional repression and participates in X chromosome inactivation of female mammals. It is involved in the initiation of both imprinted and random X inactivation. Ubiquitinated H2A is enriched in inactive X chromosome chromatin. Ubiquitination of H2A functions downstream of methylation of 'Lys-27' of histone H3 (H3K27me). H2AK119Ub by RNF2/RING2 can also be induced by ultraviolet and may be involved in DNA repair. Following DNA double-strand breaks (DSBs), it is ubiquitinated through 'Lys-63' linkage of ubiquitin moieties by the E2 ligase UBE2N and the E3 ligases RNF8 and RNF168, leading to the recruitment of repair proteins to sites of DNA damage. Ubiquitination at Lys-14 and Lys- 16 (H2AK13Ub and H2AK15Ub, respectively) in response to DNA damage is initiated by RNF168 that mediates monoubiquitination at these 2 sites, and 'Lys-63'-linked ubiquitin are then conjugated to monoubiquitin; RNF8 is able to extend 'Lys-63'-linked ubiquitin chains in vitro. H2AK119Ub and ionizing radiation-induced 'Lys- 63'-linked ubiquitination (H2AK13Ub and H2AK15Ub) are distinct events. PTM: Phosphorylation on Ser-2 is enhanced during mitosis. Phosphorylation on Ser-2 by RPS6KA5/MSK1 directly represses transcription. Acetylation of H3 inhibits Ser-2 phosphorylation by RPS6KA5/MSK1. PTM: Symmetric dimethylation on Arg-4 by the PRDM1/PRMT5 complex may play a crucial role in the germ-cell lineage (By similarity). PTM: Crotonylation (Kcr) is specifically present in male germ cells and marks testis-specific genes in post-meiotic cells, including X-linked genes that escape sex chromosome inactivation in haploid cells. Crotonylation marks active promoters and enhancers and confers resistance to transcriptional repressors. It is also associated with post-meiotically activated genes on autosomes. MASS SPECTROMETRY: Mass=13838.7; Method=Electrospray; Range=2-128; Note=Monoisotopic with N-acetylserine; Source=PubMed:16457589; SIMILARITY: Belongs to the histone H2A family.
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
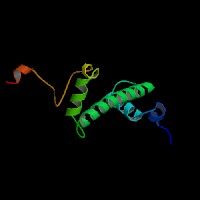
ModBase Predicted Comparative 3D Structure on Q99878
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.
Mouse
Rat
Zebrafish
D. melanogaster
C. elegans
S. cerevisiae
No ortholog
No ortholog
No ortholog
No ortholog
No ortholog
No ortholog
Gene Ontology (GO) Annotations with Structured Vocabulary