Human Gene OSTM1 (ENST00000492130.2) from GENCODE V44
Description: Required for osteoclast and melanocyte maturation and function (By similarity). (from UniProt Q86WC4) RefSeq Summary (NM_014028): This gene encodes a protein that may be involved in the degradation of G proteins via the ubiquitin-dependent proteasome pathway. The encoded protein binds to members of subfamily A of the regulator of the G-protein signaling (RGS) family through an N-terminal leucine-rich region. This protein also has a central RING finger-like domain and E3 ubiquitin ligase activity. This protein is highly conserved from flies to humans. Defects in this gene may cause the autosomal recessive, infantile malignant form of osteopetrosis. [provided by RefSeq, Jul 2008]. Gencode Transcript: ENST00000492130.2 Gencode Gene: ENSG00000081087.16 Transcript (Including UTRs) Position: hg38 chr6:108,041,409-108,074,797 Size: 33,389 Total Exon Count: 7 Strand: - Coding Region Position: hg38 chr6:108,044,785-108,074,651 Size: 29,867 Coding Exon Count: 6
ID:OSTM1_HUMAN DESCRIPTION: RecName: Full=Osteopetrosis-associated transmembrane protein 1; AltName: Full=Chloride channel 7 beta subunit; Flags: Precursor; FUNCTION: Required for osteoclast and melanocyte maturation and function (By similarity). SUBUNIT: Chloride channel 7 are heteromers of alpha (CLCN7) and beta (OSTM1) subunits. SUBCELLULAR LOCATION: Lysosome membrane; Single-pass type I membrane protein. Note=Requires CLCN7 to travel to lysosomes. PTM: Undergoes proteolytic cleavage in the luminal domain, the cleaved fragments might be linked by disulfide bonds with the remnant of the protein (By similarity). PTM: Highly N-glycosylated. DISEASE: Defects in OSTM1 are the cause of osteopetrosis autosomal recessive type 5 (OPTB5) [MIM:259720]; also called infantile malignant osteopetrosis 3. Osteopetrosis is a rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. The disorder occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. OPTB5 patients manifest primary central nervous system involvement in addition to the classical stigmata of severe bone sclerosis, growth failure, anemia, thrombocytopenia and visual impairment with optic atrophy. SEQUENCE CAUTION: Sequence=AAD27000.1; Type=Frameshift; Positions=221; WEB RESOURCE: Name=GeneReviews; URL="http://www.ncbi.nlm.nih.gov/sites/GeneTests/lab/gene/OSTM1";
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
Pfam Domains: PF09777 - Osteopetrosis-associated transmembrane protein 1 precursor
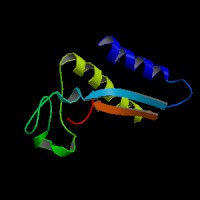
ModBase Predicted Comparative 3D Structure on Q86WC4
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.