Frequently Asked Questions: Data and Downloads
Topics
- Downloading sequence and annotation data
- Metadata tables for GenBank and RefSeq moved to hgFixed database
- Extracting sequence in batch from an assembly
- Downloading data from the UCSC DAS server
- Downloading the UCSC Genome Browser source
- Download restrictions
- Opening .fa files
- Data differences between downloaded data and browser display
- Strange characters in FASTA file
- Selection of GenBank ESTs
- EST strand direction
- Missing RefSeq ID
- Finished vs. draft segments
- chr_alt Chromosome
- chr_fix Chromosome
- chrN_random tables
- Chromosome Un
- Chromosome M
- N characters at beginning of human chr22
- Erroneous duplicated chrY_random region on Mouse Build 34 (mm6)
- Mapping chimp chromosome numbers to human chromosomes numbers
- Converting genome coordinates between assemblies
- Linking gene name with accession number
- Obtaining a list of Known Genes
- Filtering for a transcription factor in the JASPAR database
- Repeat-masking data
- Availability of repeat-masked data
- RepeatMasker version differences - UCSC vs. Repeatmasker website
- Obtaining promoter sequence
- Data from Evolutionary Conservation Score tracks
- Minus strand coordinates - axtNet files
- Mapping UCSC STS marker IDS to those of other groups
- deCODE map data
- Direct MariaDB (MySQL) access to data
- Name of fourth column in BED output
- Track data access
- How do I download dbSNP data?
- Why doesn't this SNP have two alleles?
- Known issues with Table Browser GTF output
- Table Browser output file not ordered
- 'Permission denied' error when trying to use command-line utilities
- Restricted Track Data
- What is the genome analysis set?
- How do I download GenArk data?
- Why are the conservation scores different from the ones in the download file?
- How to make programmatic queries to the Genome Browser
Return to FAQ Table of Contents
Downloading sequence and annotation data
How do I obtain the sequence and/or annotation data for a release?
Sequence and annotation data downloads are usually made available within the first week of the release of a new assembly. The download directories are automatically updated nightly to incorporate additions and modifications to the data.
You can download sequence and annotation data using our FTP server, but we recommend using rsync, which has the advantage of starting up where it left off after a failure, when run again. Please see the previous link for examples.
You can also download data from our Downloads page or our DAS server. To download a specific subset of the data or to configure the output format of the data, use the Table Browser. For information on extracting a large set of sequences from an assembly, see Extracting sequence in batch from an assembly.
For more information on using the UCSC DAS server, see Downloading data from the UCSC DAS server.
Another option for querying sequence and annotation data is the REST API. This interface allows for extraction of sequence and annotations from both UCSC assemblies and from hubs.
To quickly download large volumes of data you can use UDR (UDT Enabled Rsync): UDR provides users much faster download rates. Here is an example using UDR, once installed, to download all the mouse mm9 ENCODE information that amounts to several terabytes:
$ udr rsync -avP hgdownload.gi.ucsc.edu::goldenPath/mm9/encodeDCC/ /my/local/mm9/Optional: download from our secondary download server.
$ udr rsync -avP hgdownload2.gi.ucsc.edu::goldenPath/mm9/encodeDCC/ /my/local/mm9/Metadata tables for GenBank and RefSeq moved to hgFixed database
I can no longer find metadata tables like gbCdnaInfo for an assembly.
As of June 2016, the location of metadata tables that support the GenBank and RefSeq tracks (RefSeq, Other RefSeq, mRNA, EST, etc.) have been moved from directories of individual assemblies to one global database, hgFixed.
The tables below (previously found per assembly) can now be downloaded from the hgFixed database:
- author
- cds
- cell
- description
- development
- gbCdnaInfo
- gbExtFile
- gbLoaded
- gbMiscDiff
- gbSeq
- gbWarn
- geneName
- imageClone
- keyword
- library
- mrnaClone
- organism
- productName
- refLink
- refSeqStatus
- refSeqSummary
- sex
- source
- tissue
These tables are also accessible from:
- The Table Browser, as connected tables and joined fields described when clicking the "data format description " button
- One of our two public access MariaDB servers in the US and Europe
Extracting sequence in batch from an assembly
I have a lot of coordinates for an assembly and want to extract the corresponding sequences. What is the best way to proceed?
There are two ways to extract genomic sequence in batch from an assembly:
A. Download the appropriate fasta files from our ftp server and extract sequence data using your own tools or the tools from our source tree. This is the recommended method when you have very large sequence datasets or will be extracting data frequently. Sequence data for most assemblies is located in the assembly's "chromosomes" subdirectory on the downloads server. For example, the sequence for human assembly hg17 can be found in ftp://hgdownload.gi.ucsc.edu/goldenPath/hg17/chromosomes/. You'll find instructions for obtaining our source programs and utilities here. Some programs that you may find useful are nibFrag and twoBitToFa, as well as other fa* programs. To obtain usage information about most programs, execute it without arguments.
B. Use the Table browser to extract sequence. This is a convenient way to obtain small amounts of sequence.
- Create a custom track of the genomic coordinates in BED format and upload into the Genome Browser.
- Select the custom track in the Table browser, then select the "sequence" output format to retrieve data. We recommend that you save the file locally as gzip.
Downloading data from the UCSC DAS server
How do I download data using the UCSC DAS server?
The UCSC DAS server provides access to genome annotation data for all current assemblies featured in the Genome Browser. To view a list of the assemblies available from the DAS server and their base URLs, see http://genome.ucsc.edu/cgi-bin/das/dsn.
To construct a DAS query, combine an assembly's base URL with the sequence entry point and type specifiers available for that assembly. The entry point specifies chromosome position, and the type indicates the annotation table requested. You can view the lists of entry points and types available for an assembly with requests of the form:
http://genome.ucsc.edu/cgi-bin/das/[db_name]/entry_points
http://genome.ucsc.edu/cgi-bin/das/[db_name]/types where [db_name] is the UCSC name for the assembly, e.g. hg16, mm4.
For example, here is a query that returns all the records in the refGene table for the chromosome position chr1:1-100000 on the hg16 assembly:
http://genome.ucsc.edu/cgi-bin/das/hg16/features?segment=1:1,100000;type=refGene
For more information on DAS, see the Biodas website and the DAS specification.
A more recent alternative to the DAS server is the REST API.
Downloading the UCSC Genome Browser source
Where can I download the Genome Browser source code and executables?
The Genome Browser source code and executables are freely available for academic, nonprofit, and personal use (see Licensing the Genome Browser or Blat for commercial licensing requirements). The latest version of the source code may be downloaded here.
See Downloading Blat source and documentation for information on Blat downloads.
Download restrictions
Do you have restrictions on the amount of downloads one can do?
Generally, we'd prefer that you not hit our interactive site with programs, unless they are themselves front ends for interactive sites. We can handle the traffic from all the clicks that biologists are likely to generate, but not from programs. Program-driven use is limited to a maximum of one hit every 15 seconds and no more than 5,000 hits per day.
If you need to run batch Blat jobs, see Downloading Blat source and documentation for a copy of Blat you can run locally.
Opening .fa files
I am trying to look at the final decoding of the human genome. How can I open the *.fa files?
Microsoft Word or any program that can handle large text files will do. Some of the chromosomes begin with long blocks of Ns. You may want to search for an A to get past them.
Unless you have a particular need to view or use the raw data files, you might find it more interesting to look at the data using the Genome Browser. Type the name of a gene in which you're interested into the position box (or use the default position), then click the submit button. In the resulting Genome Browser display, click the DNA link on the menu bar at the top of the page. Select the Extended case/color options button at the bottom of the next page. Now you can color the DNA sequence to display which portions are repeats, known genes, genetic markers, etc.
Data differences between downloaded data and browser display
I downloaded the genome annotations from your MariaDB database tables, but the mRNA locations didn't match what was showing in the Genome Browser. Shouldn't they be in synch?
Yes. The Genome Browser and Table Browser are both driven by the same underlying MariaDB database. Check that your downloaded tables are from the same assembly version as the one you are viewing in the Genome Browser. If the assembly dates don't match, the coordinates of the data within the tables may differ. In a very rare instance, you could also be affected by the brief lag time between the update of the live databases underlying the Genome Browser and the time it takes for text dumps of these databases to become available in the downloads directory.
Strange characters in FASTA file
I noticed several characters other than A, C, G, T, and N in my fasta file, for example y, k, s, etc. Is the file corrupted or are these characters valid?
The characters most commonly seen in sequence are A, C, G, T, and N, but there are several other valid characters that are used in clones to indicate ambiguity about the identity of certain bases in the sequence. It's not uncommon to see these "wobble" codes at polymorphic positions in DNA sequences. The following chart (IUPAC-IUB Symbols for Nucleotide Nomenclature: Cornish-Bowden (1985). Nucl. Acids Res. 13:3021-3030) lists nucleotide symbols, including those used for ambiguity:
-------------------------------------- Symbol Meaning Nucleic Acid -------------------------------------- A A Adenine C C Cytosine G G Guanine T T Thymine U U Uracil M A or C R A or G Purine W A or T S C or G Y C or T Pyrimidine K G or T V A or C or G H A or C or T D A or G or T B C or G or T X G or A or T or C N G or A or T or C
Selection of GenBank ESTs
I am interested in ESTs. How do you select which ones from GenBank to display in the Genome Browser?
All ESTs in GenBank on the date of the track data freeze for the given organism are used - none are discarded. When two ESTs have identical sequences, both are retained because this can be significant corroboration of a splice site.
ESTs are aligned against the genome using the Blat program. When a single EST aligns in multiple places, the alignment having the highest base identity is found. Only alignments that have a base identity level within a selected percentage of the best are kept. Alignments must also have a minimum base identity to be kept. For more information on the selection criteria specific to each organism, consult the description page accompanying the EST track for that organism.
The maximum intron length allowed by Blat is 500,000 bases, which may eliminate some ESTs with very long introns that might otherwise align. If an EST aligns non-contiguously (i.e. an intron has been spliced out), it is also a candidate for the Spliced EST track, provided it meets various quality controls for intron and exon length and match quality. Start and stop coordinates of each alignment block are available from the appropriate table within the Table Browser.
Note that only 250 EST tracks can be viewed at a time within the browser. If more than 250 tracks exist for the selected region, the display defaults to a denser display mode to prevent the user's web browser from being overloaded. You can restore the EST track display to a fuller display mode by zooming in on the chromosomal range or by using the EST track filter to restrict the number of tracks displayed.
For tracks such as Non[Organism] ESTs and Non[Organism] mRNAs, some selection is done on the full set at GenBank. If a sequence is too divergent from the organism's genome to generate a significant Blat hit, it is not included in the track.
EST strand direction
Could you help me with my interpretation of EST data? If the EST is taken from the minus (-)
strand, does this always mean that the transcript is generated on the minus strand? Are two
corresponding ESTs that are assigned - and + always complementary?
I want to confirm the strand assignment for two human ESTs:
-
BQ016549 (chr22:22,310,674-22,332,143 on hg18): + strand in text and - strand in graphical
display
-
AA928010 (chr22:20,345,264-20,354,528 on hg18): - strand in text and + strand in graphical
display
The graphical display goes with the orientation of the gene in that location.
From the examples above, it can be seen that the strand to which an EST aligns is not necessarily reflected in the direction of transcription shown by the arrows in the display. When UCSC downloads mRNAs and ESTs from GenBank and aligns them to a genome assembly using Blat, each EST aligns to the + or - strand (forward or reverse direction) of the genome, which we record as + or - in the strand field of the corresponding database table, e.g. all_ests or chrN_est. The strand information (+/-) therefore indicates the direction of the match between the EST and the matching genomic sequence. It bears no relationship to the direction of transcription of the RNA with which it might be associated. Determining the direction of transcription for ESTs is not an easy task so we do some calculations to make the best guess for the transcription direction.
ESTs are sequenced from either the 5' or the 3' end. When sequenced from the 5' end, the resulting sequence is the same as that of the mRNA which it represents. With a 3' end read, the resulting sequence matches the opposite strand of the cDNA clone. Therefore, it is the reverse complement of the actual mRNA sequence. A problem occurs if the EST contributor reverse-complements the 3'-read sequence before depositing it into GenBank, with the idea that people will want the mRNA (transcription-direction) sequence. It is not always possible to determine if this has been done. Therefore, we do some calculations to try to determine the correct direction of transcription for the EST sequence.
If an EST alignment produces canonical introns (with gt-ag splice-site pairs), this is used to determine the transcription direction. For example when an EST is aligned to the genome, a canonical intron would look like this:
NNNNexonNNNNgtnnnnintronnnnnnnnagNNNNexon
Here, the two nucleotides on either end of the intron show the canonical gt-ag splice site pairs. To find transcription direction, we use a method that relies on finding gt-ag canonical pairs in one direction more often than in the opposite direction. The calculation is:
gt/ag introns minus ct/ac introns = intronOrientation
The sign of this calculated intronOrientation field (stored in the estOrientInfo table) shows the orientation of the transcript relative to the EST. Therefore, if intronOrientation is positive, then the EST appears in the display with the arrows pointing in the same direction as the EST.
Missing RefSeq ID
Why isn't my refseq ID in your database?
It may have been added after we last downloaded data from GenBank, or it may have been replaced or removed. You can check the submission date and status of an accession on the NCBI Entrez Nucleotide site.
Finished vs. draft segments
Do chrN.fa tables contain both finished and draft segments? If so, how do you determine which segments are finished?
Yes, these tables contain both finished and draft segments. Use the corresponding chrN_gold table to look them up. The quality of the draft varies. In general, the larger the contig it is in, the better the quality. The quality of the last 500 bases on either end of a contig tends to be lower than that of the rest of the contig.
How do you determine the accuracy? The base-calling program Phred analyzes the traces from the sequencing machines and assigns a quality score to these. These quality scores are used by the Phrap assembly program, which gives quality scores for the bases on the assembly as well.
chr_alt chromosomes
What is chr_alt?
The chr_alt chromosomes, such as chr5_KI270794v1_alt, are alternative sequences that differ from the reference genome currently available for a few assemblies including danRer11, mm10, hg19, and hg38. These are regions of the genome that exhibit sufficient variability to prevent adequate representation by a single sequence. UCSC labels these haplotype sequences by appending "_alt" to their names. These alternative loci scaffolds (such as KI270794.1 in the hg38 assembly, referenced as chr5_KI270794v1_alt in the browser), are mapped to the genome and provide supplemental genomic information on these variable locations. To find the regions these alternate sequences correspond to in the genome you may use the Alt Haplotypes track if one is available.
Additional information on alternative loci can be found on our hg38 patches blog post as well as the Genome Reference Consortium (GRC) website.
chr_fix chromosomes
What is chr_fix?
The chr_fix chromosomes, such as chr1_KN538361v1_fix, are fix patches currently available for the mm10, hg19, and hg38 assemblies that represent changes to the existing sequence. These are generally error corrections (such as base changes, component replacements/updates, switch point updates or tiling path changes) or assembly improvements (such as extension of sequence into gaps). These fix patch scaffold sequences are given chromosome context through alignments to the corresponding chromosome regions. A list of all chromosomes including chr_fix sequences can be found in the mm10, hg19, or hg38 assembly sequences pages.
More information on these patch sequences can be found on our hg38 patches blog post as well as on the the Genome Reference Consortium (GRC) website.
chrN_random tables
What are the chrN_random_[table] files in the human assembly? Why are they called random? Is there something biologically random about the sequence in these tables or are they just not placed within their given chromosomes?
In the past, these tables contained data related to sequence that is known to be in a particular chromosome, but could not be reliably ordered within the current sequence.
Starting with the Apr. 2003 human assembly, these tables also include data for sequence that is not in a finished state, but whose location in the chromosome is known, in addition to the unordered sequence. Because this sequence is not quite finished, it could not be included in the main "finished" ordered and oriented section of the chromosome.
Also, in a very few cases in the Apr. 2003 assembly, the random files contain data related to sequence for alternative haplotypes. This is present primarily in chr6, where we have included two alternative versions of the MHC region in chr6_random. There are a few clones in other chromosomes that also correspond to a different haplotype. Because the primary reference sequence can only display a single haplotype, these alternatives were included in random files. In subsequent assemblies, these regions have been moved into separate files (e.g. chr6_hla_hap1).
Chromosome Un
What is chrUn?
ChrUn contains clone contigs that cannot be confidently placed on a specific chromosome. For the chrN_random and chrUn_random files, we essentially just concatenate together all the contigs into short pseudo-chromosomes. The coordinates of these are fairly arbitrary, although the relative positions of the coordinates are good within a contig. You can find more information about the data organization and format on the Data Organization and Format page.
Chromosome M
What is chromosome M (chrM)?
Mitochondrial DNA.
N characters at beginning of human chr22
When I download human chr22 from your web site, the unzipped file contains only Ns.
There is a large block of Ns at the beginning and end of chr22. Search for an A to bypass the initial group of Ns.
Erroneous duplicated chrY_random region on Mouse Build 34 (mm6)
On the mm6 assembly, I've found duplicate contigs that are placed on both chrY and chrY_random. Is this intentional?
On the mm6 assembly, chrY_random erroneously contains a region duplicated from chrY. Because NCBI discovered this assembly problem after the UCSC Genome Browser was processed, we were not able to remove it from mm6 prior to the browser's release. The duplicated section occupies chrY:1-696,521 and chrY_random:29,615,053-30,311,573 (the end of the chromosome) and includes the following repeated fragments:
- AC134433.3
- AC145392.2
- AC148319.2
- AC145571.3
- AC145393.4
The fragments are assembled into the contig NT_111995 for chrY_random and also appear (under different names) as regions on contigs MmY_110865_34, MmY_78990_34 and NT_078925.
Mapping chimp chromosome numbers to human chromosomes numbers
How do the chimp and human chromosome numbering schemes compare?
The following table shows the mapping of chromosomes in the chimp draft assemblies to human chromosomes. Starting with the panTro2 assembly, the numbering scheme was changed to reflect a new standard that preserves orthology with human chromosomes. Initially proposed by E.H. McConkey in 2004, the new numbering convention was subsequently endorsed by the International Chimpanzee Sequencing and Analysis Consortium. This standard assigns the identifiers "2a" and "2b" to the two chimp chromosomes that fused in the human genome to form chromosome 2 and renumbers the other chromosomes to more closely match their human counterparts. As a result, chromosomes 2 and 23 (present in the panTro1 assembly) do not exist in later versions.
| Human Chr | Chimp Chr (panTro1) | Chimp Chr (panTro2) |
|---|---|---|
| 1 | 1 | 1 |
| 2 (part) | 12 | 2a |
| 2 (part) | 13 | 2b |
| 3 | 2 | 3 |
| 4 | 3 | 4 |
| 5 | 4 | 5 |
| 6 | 5 | 6 |
| 7 | 6 | 7 |
| 8 | 7 | 8 |
| 9 | 11 | 9 |
| 10 | 8 | 10 |
| 11 | 9 | 11 |
| 12 | 10 | 12 |
| 13 | 14 | 13 |
| 14 | 15 | 14 |
| 15 | 16 | 15 |
| 16 | 18 | 16 |
| 17 | 19 | 17 |
| 18 | 17 | 18 |
| 19 | 20 | 19 |
| 20 | 21 | 20 |
| 21 | 22 | 21 |
| 22 | 23 | 22 |
| X | X | X |
| Y | Y | Y |
Converting genome coordinates between assemblies
I've been researching a specific area of the human genome on the current assembly, and now you've just released a new version. Is there an easy way to locate my area of interest on the new assembly?
You can migrate sequences from one assembly to another by using the Blat alignment tool or by converting assembly coordinates. There are two conversion tools available on the Genome Browser web site: the Convert utility and the LiftOver tool. The Convert utility, which is accessed from the View menu on the Genome Browser annotation tracks page, supports forward, reverse, and cross-species conversions, but does not accept batch input. The LiftOver tool, accessed via the Tools link on the Genome Browser home page, also supports forward, reverse, and cross-species conversions, as well as batch conversions.
Note: It is not recommended to use LiftOver to convert SNPs between assemblies, and more information about how to convert SNPs between assemblies can be found on the following FAQ entry.
If you wish to update a large number of coordinates to a different assembly and have access to a Linux platform, you may find it useful to try the command-line version of the LiftOver tool. The executable file for this utility can be downloaded here. LiftOver requires a pre-generated over.chain file as input, available for selected assemblies from the Downloads page. If the desired file is not available, send a request to the genome mailing list and we may be able to provide you with one.
Using liftOver
Here is an example on how to set up and run LiftOver from the command line:
- Download the LiftOver program for your computer's operating system here
- Change permissions on that file so that it can be executed
chmod +x liftOver
- Run the program with no arguments to see the usage statement
./liftOver
liftOver - Move annotations from one assembly to another usage: liftOver oldFile map.chain newFile unMapped ...
- Download your genome conversion chain file from the
downloads directory.
For example, the human to mouse conversion (hg38ToMm10) can be downloaded like so:
wget http://hgdownload.gi.ucsc.edu/goldenPath/hg38/liftOver/hg38ToMm10.over.chain.gz
- Prepare your BED file input. Here is a few lines from a BED file you can
copy into a text file, saved as "preLift.bed".
chr1 11166587 11191615 MTOR chr9 136130562 136150630 ABO chr12 25358179 25403854 KRAS chrX 151335633 151619831 GABRA3
- You can now use the following command to LiftOver a BED file with annotations in your original
genome, "preLift.bed", with your successful conversions in "conversions.bed" and
unsuccessful conversions in "unMapped".
./liftOver preLift.bed hg19ToHg38.over.chain.gz conversions.bed unMapped
Linking gene name with accession number
I have the accession number for a gene and would like to link it to the gene name. Is there a table that shows both pieces of information?
If you are looking at the RefSeq Genes, the refFlat table contains both the gene name (usually a HUGO Gene Nomenclature Committee ID) and its accession number. For the Known Genes, use the kgAlias table.
Obtaining a list of Known Genes
How can I obtain a complete list of all the genes in the UCSC Known Genes table for a particular organism?
To obtain a complete copy of the entire Known Genes data set for an organism, open the Genome Browser Downloads page, jump to the section specific to the organism, click the Annotation database link in that section, then click the link for the knownGene.txt.gz table.
Data for a specific region or chromosome may be obtained from the Table Browser by selecting the "Genes and Gene Prediction Tracks" group, the "UCSC Genes" track and the "knownGene" table. Set the position to the region of interest, then click the "get output" button.
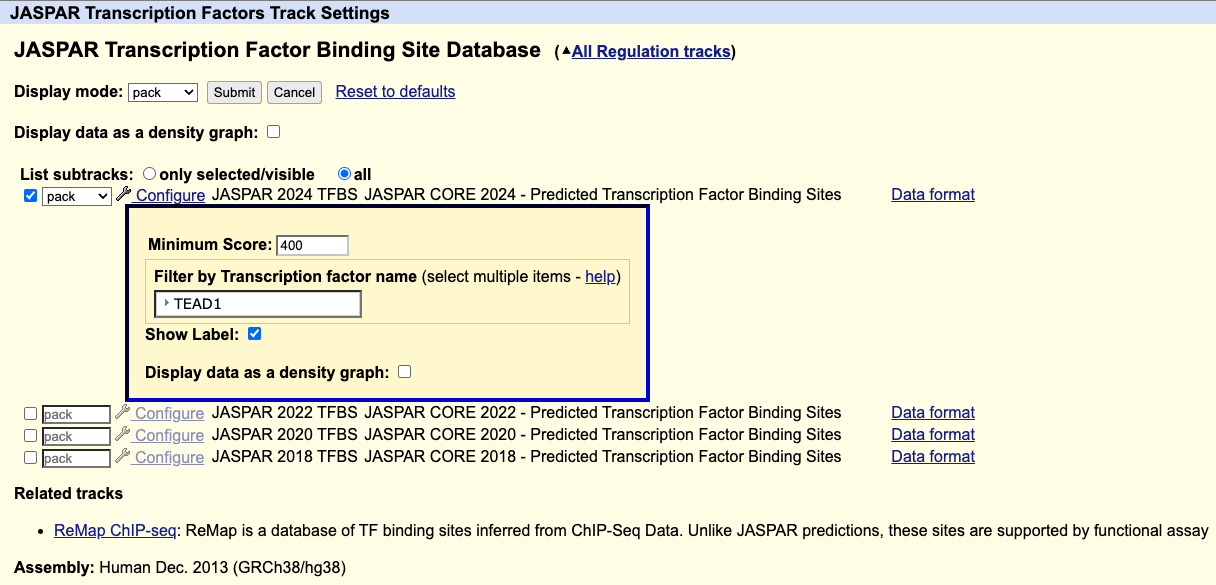
Filtering for a transcription factor in the JASPAR database
How do I display only one transcription factor?
On the track settings page for the JASPAR Transcription Factors track, you can filter for a transcription factor using the Filter by Transcription factor name setting.
For example, if you wanted to only display the TEAD1 transcription factor, under "Filter by Transcription factor name" select TEAD1, then click .
How can I download the data for only one transcription factor using the Table Browser?
On the Table Browser, you can create a filter to limit the output to a set of transcription factors. A summary of the steps is as follows:
- Select a human or mouse genome with the JASPAR track, e.g. hg38 or mm10.
- Select the JASPAR Transcription Factors track.
- Define the region with the position field. This track is unavailable for genome-wide download as the connection will timeout due to the billions of items in the track.
- Filter for the transcription factors by clicking
- On the resulting page, create a filter similar to an SQL query:
TFName does match [transcription_factor]
e.g.TFName does match TEAD1 - Click
- On the resulting page, create a filter similar to an SQL query:
- Select the output format and then click

Repeat-masking data
What version of RepeatMasker do you use on your data? Which flags do you use?
UCSC uses the latest versions of RepeatMasker and repeat libraries available on the date when the assembly data is processed. RepeatMasker version information can usually be found in the README text for the assembly's bigZips downloads directory.
Masking is done using the RepeatMasker -s flag. For mouse repeats, we also use -m. In addition to RepeatMasker, we use the Tandem Repeat Finder (trf) program, masking out repeats of period 12 or less. The repeats are just "soft" masked. Alignments are allowed to extend through repeats, but not initiate in them.
Availability of repeat-masked data
Are the repeat annotation files available for every chromosome?
Yes, you can obtain the repeat-masked files via the Table Browser or from the organism's annotation database downloads directory. The RepeatMasker annotation tables are named chrN_rmsk (where N represents the chromosome number) and the Tandem Repeat Finder (TRF) tables are named simpleRepeat.
RepeatMasker version differences - UCSC vs. RepeatMasker website
When I run RepeatMasker independently from the RepeatMasker web server, my results vary from those of UCSC. What's the cause?
UCSC occasionally uses updated versions of the RepeatMasker software and repeat libraries that are not yet available on the RepeatMasker website (see Repeat-masking data for more information).
Obtaining promoter sequence
How can I fetch promoter sequence upstream of a gene?
The UCSC Genome Browser offers several ways to obtain this information, depending on your requirements.
The Genome Browser downloads site provides prepackaged downloads of 1000 bp, 2000 bp, and 5000 bp upstream sequence for RefSeq genes that have a coding portion and annotated 5' and 3' UTRs. You can obtain these from the bigZips downloads directory for the assembly of interest.
To fetch the upstream sequence for a specific gene, use the Table Browser. Enter the genome, assembly, and select the knownGene table. Paste the gene name or accession number in the identifier field. Choose sequence for the output format type, then click the get output button. On the next page, select genomic. On the final page, you will have the opportunity to configure the amount of upstream promoter sequence to fetch, along with several other options. Click Get Sequence when you've finished configuring the output.
You can also use the Genome Browser to obtain sequence for a specific gene. Open the Genome Browser window to display the gene in which you're interested. Click the entry for the gene in the RefSeq or Known Genes track, then click the Genomic Sequence link. Alternatively, you can click the DNA link in the top menu bar of the Genome Browser tracks window to access options for displaying the sequence.
The Stanford Human Promoters track on the UCSC Custom Annotation Tracks page shows promoters for some of the human assemblies.
Data from Evolutionary Conservation Score tracks
Where can I download the conservation score data from the Human/Mouse Evolutionary Conservation Score track?
The conservation score data are stored in a group of tables in the annotation database downloads directory. The naming conventions of the tables vary among releases. In earlier assemblies, table names are of the form chrN_humMusL, chrN_zoom1_humMusL, and or chrN_zoom2500_humMusL. In later releases, the tables are named using specific release numbers, such as chrN_hg16Mm3. The tables within a given set differ by the number of bases/score interval and are used to generate the browser displays at different zooming levels.
Minus strand coordinates - axtNet
I downloaded the axtNet alignments between the latest human and mouse assemblies. I found that some of the alignments listed in the axtNet files do not agree with what is shown in the browser.
Is this alignment on the minus strand? Minus strand coordinates in axt files are handled differently from how they are handled in the Genome Browser. To convert axt minus strand coordinates to Genome Browser coordinates, use:
start = chromSize + 1 - axtEnd end = chromSize + 1 - axtStart
See an explanation of coordinate transforms in the genomeWiki.
Mapping UCSC STS marker IDs to those of other groups
How do I map the STS genetic marker IDs in the genome browser to the IDs assigned by other groups?
We assign our own IDs to each of the STS markers, but we also track the UniSTS IDs for each marker in the downloadable stsInfo2 table. To determine the location of a specific marker, look up the marker's name in the stsAlias table to determine the UCSC ID assigned to the marker, and then use this ID to look it up in the stsMap table where the marker is located. For example, D10S249 has UCSC ID 2880 and is located at chr10:240791-241019.
deCODE map data
Where can I get more information about the deCODE map?
You can obtain this information from the combination of a couple of tables. The stsMap table contains the physical position of all STS markers, including those on the deCODE map. This file also contains information about the position on the genome-wide maps, including the deCODE map. A second file, stsInfo2, contains additional information about each marker, including aliases, primer sequence information, etc. This table is related to the first table by an ID (the identNo field in both files).
Direct MariaDB (MySQL) access to data
Is it possible to run SQL queries directly on the database rather than using the Table Browser interface?
Yes. See our documentation on Downloading Data using MariaDB (MySQL).
Connect to the US MariaDB server using the command:
mysql --user=genome --host=genome-mysql.gi.ucsc.edu -A Or to the European MariaDB server using the command:
mysql --user=genome --host=genome-euro-mysql.soe.ucsc.edu -A Name of fourth column in BED output
When using the Table Browser to extract exons from a Gene track, what does the "Name" column (fourth BED column) refer to?
The fourth column of the BED output contains a lot of information separated by underscores. For example:
uc009vjk.2_cds_1_0_chr1_324343_f This information is represented as follows:
ucscId_sequenceType_sequenceTypeNumber_basesAdded_chromosome_positionOfFirstBaseOfItem_strand- UCSC ID: our identification for the transcripts in the UCSC Genes track.
- Sequence Type: exons, introns, cds, utr5, etc.
- Sequence Type Number: for every transcript, there will be a row for each sequence type (cds or intron) and this identifies which is represented in this row; the first is denoted with 0. So, if you requested exons, and a particular transcript has 10 exons, you will see a row for each one and in this position they will be numbered 0-9.
- Bases Added: number of bases added to the regions requested.
- Chromosome: chromosome number the item is on.
- Position of First Base of Item: if you have specified bases added to the requested features (for example, Exons plus 10 bases on each end), then columns 2 and 3 of the output wouldn't be the exact coordinates of the exon, they would start and end 10 bases before/after the exon. So, this part of the information is an easy way to see where the actual feature starts as displayed in the browser. It is "as displayed in the browser" because the coordinates in our tables almost always have 0-based starts (as they do in columns 2 and 3 of this output) but display as 1-based in the browser (for more info see this FAQ), but this start position listed in this section of the 4th column is actually 1 based. It will be the exact coordinate the feature starts on as displayed in the browser.
- Strand: forward(f) or reverse(-) strand.
Track Data Access
How do I access the data underlying a track?
The raw data underlying a track can be explored interactively with the Table Browser, Data Integrator, or Variant Annotation Integrator. For automated analysis, the genome annotation can be downloaded from the downloads server, one of our two public MariaDB servers, or using our REST API.
bigBed data: For bigBed files, individual regions or the whole genome annotation can be obtained using our tool bigBedToBed which can be compiled from the source code or downloaded as a precompiled binary for your system. Instructions for downloading source code and binaries can be found here. The tool can also be used to obtain only features within a given range using our hgdownload server, example:
-
North American server:
bigBedToBed http://hgdownload.gi.ucsc.edu/gbdb/path/to/file/bigBedfile.bb -chrom=chr21 -start=0 -end=1000000 stdout
Read more in our blog about Accessing the Genome Browser Programmatically to acquire data.
How do I download dbSNP data?
For versions dbSNP153 and above, the data is formatted in bigBed files. Previous versions are MySQL tables. For help with versions before dbSNP153, see accessing MySQL data. This FAQ entry pertains to versions dbSNP153 and above.
Since dbSNP has grown to include over 700 million variants, the size of the All dbSNP (153+) subtrack can cause the Table Browser and Data Integrator to time out, leading to a blank page or truncated output, unless queries are restricted to a chromosomal region or to a specific set of rs# IDs (which can be pasted/uploaded into the Table Browser), or to one of the subset tracks such as Common or ClinVar.
For automated analysis, the track data files can be downloaded from the downloads server for hg19 and hg38. Below are specific examples for dbSNP153, however, the same methods and directories will work by substituting a more recent dbSNP release.
| file | format | subtrack | ||
|---|---|---|---|---|
| dbSnp153.bb | hg19 | hg38 | bigDbSnp (bigBed4+13) | All dbSNP (153) |
| dbSnp153ClinVar.bb | hg19 | hg38 | bigDbSnp (bigBed4+13) | ClinVar dbSNP (153) |
| dbSnp153Common.bb | hg19 | hg38 | bigDbSnp (bigBed4+13) | Common dbSNP (153) |
| dbSnp153Mult.bb | hg19 | hg38 | bigDbSnp (bigBed4+13) | Mult. dbSNP (153) |
| dbSnp153BadCoords.bb | hg19 | hg38 | bigBed4 | Map Err (153) |
| dbSnp153Details.tab.gz | gzip-compressed tab-separated text | Detailed variant properties, independent of genome assembly version | ||
Several utilities for working with bigBed-formatted binary files can be downloaded here. Run a utility with no arguments in order to see a brief description of the utility and its options.
- bigBedInfo provides summary statistics about a bigBed file including the number of items in the file. With the -as option, the output includes an autoSql definition of data columns, useful for interpreting the column values.
- bigBedToBed converts the binary bigBed data to tab-separated text. Output can be restricted to a particular region by using the -chrom, -start and -end options.
- bigBedNamedItems extracts rows for one or more rs# IDs.
Example: retrieve all variants in the region chr1:200001-200400
bigBedToBed http://hgdownload.gi.ucsc.edu/gbdb/hg38/snp/dbSnp153.bb -chrom=chr1 -start=200000 -end=200400 stdout
Example: retrieve variant rs6657048
bigBedNamedItems dbSnp153.bb rs6657048 stdout
Example: retrieve all variants with rs# IDs in file myIds.txt
bigBedNamedItems -nameFile dbSnp153.bb myIds.txt dbSnp153.myIds.bed
The columns in the bigDbSnp/bigBed files and dbSnp153Details.tab.gz file are described in bigDbSnp.as and dbSnpDetails.as respectively.
UCSC has an API that can be used to retrieve values from a particular chromosome range. A list of rs# IDs can also be pasted/uploaded in the Variant Annotation Integrator tool in order to find out which genes (if any) the variants are located in, as well as functional effect such as intron, coding-synonymous, missense, frameshift, etc.
See our searchable mailing list archives for more information and example queries. We also have information on our blog about Accessing the Genome Browser Programmatically to acquire data.
Why doesn't this SNP have two alleles?
When using the SNP tracks, some records may contain information about one or more alleles instead of the usual two alleles for the SNP. The following information should explain how this is possible.
- One allele (i.e. reference only):
- The human genome reference has gone through many different assembly versions. The reference genome has always been a mosaic of sequences from multiple individuals, so it contains some rare or singleton mutations and is not entirely free of errors. Some SNPs were discovered on previous assembly versions, and the latest assembly version has the corrected or common allele, which turns out to be the only observed allele (so the SNP was an artifact of the reference assembly having a rare mutation or error in the past, not a real SNP).
- Three alleles:
- It's rare, but possible, for the same base to be mutated to different values in different people.
- Four alleles:
- This would be even rarer than three alleles. In the past, it has often been a symptom of strand errors, for example, the same variant is reported separately as A/G on the forward strand and C/T on the reverse strand, but then the strand information being lost in processing and the reports merged to A/C/G/T.
Obtaining GTF (Gene Transfer Format)
What is the best method for obtaining GTF output?
Currently, the Table Browser option return data in
GTF format is limited as explained below.
To convert custom GenePred format data into GTF, the best method is to use the
command-line format conversion utility, genePredToGtf. This can optionally be set up
to automatically connect to the UCSC public SQL database and return GTF files in a few minutes using
this short guide.
For simplicity, GTF files have been generated using the genePredToGtf method
described above and are available on our download server for the main gene transcript sets.
These can be found at the following download server address:
http://hgdownload.gi.ucsc.edu/goldenPath/$db/bigZips/genes/
where $db is the assembly of interest. For example, the hg38 GTF files.
Summary of Table Browser limitations:
- The Table Browser has transcript IDs only, so although it includes both "gene_id" and "transcript_id" fields in its output, the value for transcript ID (e.g., ENST#) is used for both fields.
- The Table Browser adds start and stop codon annotations whether or not the transcript alignment includes proper start and stop codons.
- Some tables in older genome assemblies are not supported.
GenePred (short for Gene Predictions) is a table
format commonly used for gene tracks in the UCSC Genome Browser where each transcript has a single
row. Tables are not stored in GTF as it would require many rows to describe a single transcript
since each gene feature (i.e., exon) requires a separate line. The genePredToGtf command-line
utility can be used to convert genePred to GTF. Download the genePredToGtf operating
system-specific command-line utility from the
utilities directory.
Please see the Genes in GTF
or GFF Format wiki page for examples and various methods for conversion. The genePredToGtf
utility can convert files from several sources, such as Table Browser output from a genePred table,
a local downloaded gene set table like refGene.txt, or from querying
public MariaDB tables.
Table Browser output file order
My table browser output file is not ordered by position, how is it ordered?
Most of our tables have a special first column called "bin" that helps with quickly displaying data on the Genome Browser. This (chrom,bin) index causes query results to be ordered first by bin, then by chromStart. This allows us to query and return results more quickly than if they were sorted by chromStart.
A quick way to sort an output BED file by position is to use the following UNIX command on our Table Browser output BED file:
sort -k1,1 -k2n,2n example.bed > example.sorted.bed'Permission denied' error when trying to use command-line utilities
Why do I get a 'Permission denied' error when I try to run command-line utilities?
In order for your computer to run a freshly downloaded utility, you will need to update the file
system permissions to allow your operating system to run the program.
To make utilities usable, turn on its 'executable' bit:
$ chmod +x ./filePath
$ ./filePath/utility_name
Example:
$ chmod +x /home/user/liftover/liftOverRestricted Track Data
Why can I not download some data in the Table Browser or find the download files?
Some data is provided by external groups and is not available for download or mirroring by any third party without the permission of the owners, such as the OMIM track data, which is the property of Johns Hopkins University. For some tools, such as attempting a getData fetch with our API of restricted tracks, a 403 'Forbidden' error will be returned. Please email our private internal genome-www@soe.ucsc.edu mailing list if you have any questions.
Analysis set
Some genomes in the download server also reference an analysis set, what is the difference?
For certain genomes (GRCm38/mm10, GRCh37/hg19, GRCh38/hg38), NCBI provides an analysis set in addition to the standard genome files. These are FASTA files with modified sequence identifiers and index files convenient for analysis with Next Generation Sequencing tools. These files are particularly helpful for NGS pipelines including variant calling and RNA-Seq analysis.
Though not all analysis sets contain the same information, features include:
- Removal of alternate and fix sequences which can interfere with read alignment programs
- Hard masking of duplicate copies of the pseudo-autosomal regions (PARs) and centromeric arrays
- Addition of "decoy" sequences
- Index files generated by BWA, Samtools, Bowtie and HISAT2
For more information on analysis sets, see the NCBI FAQ. Information on what is contained in each specific assembly analysis set can be found in the README by clicking the Genome sequence files link for the assembly of interest in our Downloads page.
GenArk Downloads
How do I download GenArk assembly hub data for my species?
For 2000+ GenArk genomes, we visualize them in assembly hubs instead of native assemblies like hg38 and mm39. These Genome Browsers can be accessed from our Genomes page by searching common name or GCA/GCF number. You can also access the browsers for these species directly with links in the following format:
https://genome.ucsc.edu/h/GCF_000951035.1
The downloads data for these assemblies is stored in a different location than our goldenPath, SQL, or gbdb file directories. There are two ways to access this data for download. First, you can go to the GenArk page and select your clade (primates, mammals, birds, etc.) and then you will be brought to a page with a table of species and GCA/GCF assembly identifiers. Find your genome and click on the third column, labeled "Scientific name and data download", which will take you to the download directory for that species.
Alternatively, you can enter your GCA/GCF identifier in the URL in groups of three characters, separated by slashes. For example, the identifier "GCA_004027835.1" has data in the following directory:
https://hgdownload.gi.ucsc.edu/hubs/GCA/004/027/835/
Conservation scores downloads
Why are the conservation scores on the UCSC Genome Browser site different from the ones in the download file?
The difference in the conservation scores, for both PhastCons and PhyloP, is that the wiggle database format (from which the details page and Table Browser scores are extracted) uses lossy compression that keeps enough resolution to display the pixelated scores in the browser graphic display but does not reconstruct the true original scores. This is why we make the original score files available for download.
How to make programmatic queries to the Genome Browser
To access the UCSC Genome Browser programmatically and bypass the CAPTCHA, all requests must include an API key. Note that an API key is not needed to use our REST API.
API key access is only available on our primary site (not available on genome-euro or genome-asia). To generate your own key, first log in with your UCSC Genome Browser account. Once logged in, go to the Hub Development page and generate a key from the API key section at the bottom of the page.
The key must be appended to your request URLs using the apiKey= variable.
Separate it from other URL parameters with an ampersand (&). For example, most queries will
work by adding apiKey=YOUR_UNIQUE_KEY to the end of the URL:
https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg38&apiKey=YOUR_UNIQUE_KEY
If you experience any issues using the API key, please contact us.